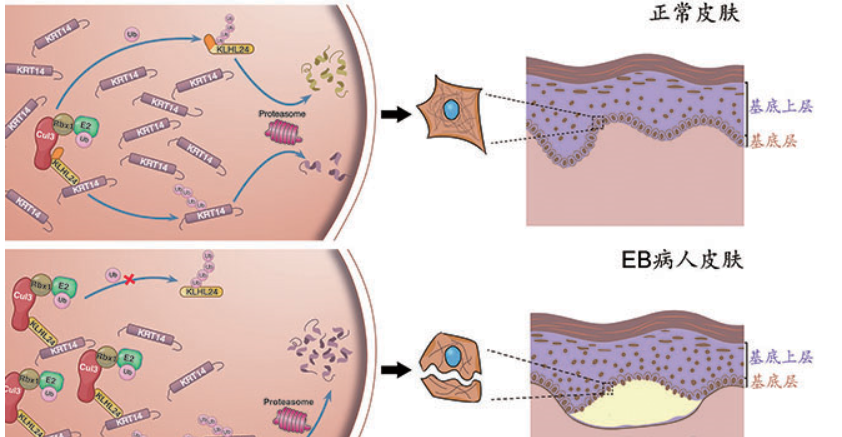
是遗传性大疱性表皮松解症(EB),一种罕见的遗传性皮肤疾病。这种病的患者被称作蝴蝶宝贝,是因为他们的皮肤和蝶翅一样脆弱。
EB是一种非常罕见的遗传性皮肤病,这种病是由于患者自身不能正常合成皮肤中所需要的结构蛋白,皮肤缺乏正常人皮肤的弹性和韧度,他们的皮肤稍微受到外力就可能在表皮和真皮层中间形成水疱,甚至溃烂。
各型大疱性表皮松解症的共同特点是皮肤在受到轻微摩擦或碰撞后出现水疱及血疱,好发于肢端及四肢关节伸侧,严重者可累及机体任何部位。皮损愈合后可形成瘢痕或粟丘疹,肢端反复发作的皮损可使指趾甲脱落。
单纯型仅累及肢端及四肢关节伸侧,不累及黏膜,皮损最表浅,愈后一般不留瘢痕。营养不良型可累及任何部位(包括黏膜),病情多较重,常在出生后即出现皮损,且位置较深,愈合后遗留明显的瘢痕,肢端反复发生的水疱及瘢痕可使指趾间的皮肤粘连,指骨萎缩形成爪形手。
口咽部黏膜反复溃破、结痂可致张口、吞咽困难,预后不佳。交界型罕见,出生后即出现广泛水疱、大疱及糜烂面,预后差,多在2岁内死亡。
EBA多发生在老年人,皮损好发生在手指、足、肘膝关节侧面,容易受外伤的部位,皮损为无炎症反应的皮肤上形成水疱、大疱、糜烂等损害,愈后可留萎缩性瘢痕,可见庆档厅粟丘疹,部分病人伴有毛发、甲损害,以及黏膜损害。
国际罕见病日
蝴蝶宝贝(遗传性大疱性表皮松解症(EB))是属于罕见病的其中一种,很多罕见病的发病率仅有十几万分之一,因其病种罕见且只有不到1%的病有治疗方法,因此罕见病也被人形象地称作“孤儿病”。目前,国内较为人熟知的罕见病包括苯丙酮尿症、地中海贫血、成骨不全症(俗称玻璃娃娃)等,但仍有很多罕见病不为人知。
建立时间:
2008年2月29日,欧洲罕见病组织(EURODIS)发起了第一届国际罕见病日。选择此时间点是由于这是每四年才出现一次的日子,寓意罕见。欧洲罕见病组织成立于1997年,是在欧盟各成员国、企业基金会、健康机构等支持下。
由罕见病患者组织及罕见病领域的爱心人士组成的非政府组织,其宗旨是改善欧洲罕见病患者的生命质量。其后在各国的一致拥护下,将每年二月的最后一天定为国际罕见病日。
至今全球已确认的罕见病有六七千种,约占人类总疾病的10%。中国罕见病涉及千万人,有人呼吁将罕见病列入社保。
起源:
国际誉隐罕见病日旨在促进社会公众和政府对罕见病及罕见病群体面临的问题的认知,罕见病,是指流行率很低、很少见的疾病,一般为慢性、严重性疾病,常危及生命。世界卫生组织将罕见病定义为患病人数占总人口的0.65‰~1‰之间的疾病或病变。
国际确认的罕见病有五六千种,约占人类疾病的10%。按此比例,我国各类罕见病患者总数应有千万人之多。但绝大部分医务人员对罕见病缺乏治疗经验和研究,对患者束手无策;多数罕见病患者对自己的疾病了解甚少,不知如何正确治疗。
病患长期在家,未能接受正常的学校教育,缺乏就业技能和独立生活的能力;部分罕见病患者家庭因病致贫,生活艰难。
在患者及其家属、爱心人士、医务人员、非政府组织、社会媒体的共同努力下,对罕见病患者的关爱活动在越来越多的国家、特别是发达国家开始进行,可以说,对罕见病患者的关注与社会的文明与进步程度密切相关。
根据世界卫生组织报道,约有80%的罕见病由于遗传缺陷引起,约有50%的罕见病在出生时或者儿童期即可发病。罕见病常进展迅速,死亡率很高,仅有约1%的蠢激罕见病有有效治疗药物。各国对罕见病认定的标准存在一定差异,这与其罕见病药物研发的激励政策及罕见病诊疗费用的覆盖范围有关。
标签:蝴蝶,宝贝